
一、研究目的
化学药品生产过程中直接接触的工艺组件,可能与液体接触并发生相互作用,导致相关浸出物的产生和积累。浸出物在液体中持续存在并最终传递至终产品中,可能影响产品质量和/或患者安全。
本方案参考《化学药品注射剂生产所用的塑料组件系统相容性研究技术指南(试行)》、《化学药品与弹性体密封件相容性研究技术指导原则(试行)》和《化学药品注射剂与塑料包装材料相容性研究技术指导原则(试行)》等相关指导原则及法规要求,对烟台鲁银药业有限公司委托的直接接触药品的工艺组件进行相容性研究,研究内容包括:可提取物研究、分析方法开发与验证、浸出物研究、安全性评估。
可提取物研究即通过选择合理的模拟提取条件对工艺组件进行提取研究,预测可能迁移到药品的目标浸出物;方法学验证是对目标浸出物的检测方法的适用性进行验证;浸出物研究是确定目标浸出物及相关方法学验证基础上,检测迁移样品中浸出物的含量,与相关阈值进行比较,初步评估药品中浸出物的安全性;对于超出阈值的物质需进行毒理学评估,以进一步评估药品与工艺组件之间的相容性。根据以上研究内容为该工艺组件是否适用于药品生产提供依据。
二、样品信息及需求量
2.1 药品信息
表1:药品信息
基本信息 | 厂家名称 | XXXXXX有限公司 | ||
药品名称 | 氯化钠注射液注射液 | |||
规格 | 1ml:20mg | 药品pH范围 | 3.7-5.5 | |
给药方式 | 肌肉注射、静脉注射、静脉滴注 | 每日最大用药量 | 5ml/100mg | |
批量 | 6万支(1ml) | 备注 | 无 | |
成分 | 类别 | 成分/含量 | ||
原料药 | 氯化钠注射液:20mg | |||
辅料 | 注射用水:1ml | 氯化钠:6mg | ||
2.2 组件信息
表2:组件信息
序号 | 生产组件名称 | 材质 | 规格 | 接触时间h | 接触温度℃ | 组件与药液接触面积cm2 | 该材质组件与药液总接触面积cm2 |
1 | 不锈钢配料罐 | 316L不锈钢 | 300L | 19 | 80℃ | 30000 | 50200 |
2 | 不锈钢配料罐 | 316L不锈钢 | 20L | 4 | 40℃ | 1600 | |
3 | 不锈钢管路 | 316L不锈钢 | N.A. | 19 | 80℃ | 11000 | |
4 | 料液接收器 | 316L不锈钢 | 20L | 11 | 40℃ | 5200 | |
5 | 冗余过滤器 | 316L不锈钢 | N.A. | 11 | 40℃ | 1100 | |
6 | 主过滤器 | 316L不锈钢 | N.A. | 11 | 40℃ | 1100 | |
7 | 针头 | 316L不锈钢 | N.A. | 11 | 40℃ | 200 | |
8 | 玻璃十通 | 玻璃 | N.A. | 11 | 40℃ | 800 | 1800 |
9 | 玻璃泵 | 玻璃 | N.A. | 11 | 40℃ | 1000 | |
10 | 硅胶管 | 硅胶 | N.A. | 11 | 40℃ | 6000 | 10000 |
11 | 硅胶垫 | 硅胶 | N.A. | 15 | 40℃ | 3000 | |
连接软管 | 硅胶 | N.A. | 19 | 40℃ | 1000 |
2.3 浸出物研究用样品
表3:迁移试验条件及样品需求量
样品需求量 | 用途 | |
正常条件生产的样品 | 1批 | 方法学开发与验证 |
正常条件生产的样品 | 3批 | 迁移试验浸出物研究 |
三、元素限度值和化合物的分析评价阈值
3.1 元素限度值
根据 ICHQ3D元素杂质指导原则,将PDE转换为浓度作评估药品或者其他组分中元素杂质含量的工具,即为元素限度值(concentration limit)。
元素限度值将参考 ICHQ3D、USP232、
3.2 化合物的分析评价阈值(AET)
阈值又叫临界值,相容性研究用化合物分析评价阈值,化合物的分析评价阈值(AET)有2种转化方法,即SCT法和PDE法,转化成AET时需结合产品的给药方式、给药方案、治疗周期等参数。
3.2.1 SCT转化为AET
SCT为安全性评估阈值(Safety Concern Threshold,SCT),当化合物低于SCT时,可认为化合物的含量非常小,其致癌性和基因毒作用可以被忽略,不会对患者产生安全性隐患,并不需要被报告,SCT与AET之间的转化计算公式为:
AET=SCT/日最大用药量×(1−50%),其中:50%为不确定度。
不同给药途径的SCT不同。目前欧洲药品局(European Medicines Agency,EMA)推荐的遗传毒性致癌物的安全性阈值(SCT)注射剂SCT为1.5μg/天,国际药用气雾剂联盟(International Pharmaceutical Aerosol Consortium,IPAC)推荐的吸入制剂SCT为0.15μg/天,注射制剂为1.5μg/天。
表4:硅胶的 AET
SCT | AET |
1.5μg/天 | 0.9μg/cm2 |
计算公式:AET(μg/cm2)=SCT(μg/天)/每日最大用药量所对应的硅胶接触面积(cm2/天)×(1-50%) 其中每日最大用药量所对应的硅胶接触面积=5(ml/天)/60000(ml)×10000 (cm2)=0.83 cm2/天 | |
3.2.2 PDE转化为AET
PDE为人每日允许暴露量,主要针对由文献、毒性数据库能获得PDE的化合物,根据制剂的临床使用情况,如:每日最大使用剂量、给药方式等,计算得到分析评价阈值(AET),与分析测得的化合物浓度进行比较,得出该浸物水平是否符合安全性要求。PDE与AET之间的转化计算公式为:
AET=PDE/日最大用药量×(1−50%),其中:50%为不确定度。
四、提取试验研究
模拟提取试验是指采用合适的模拟溶剂(如缓冲液、有机相),在较剧烈的条件下,对工艺组件材料进行的提取试验研究;目的是通过提取试验,对可提取物(工艺组件材料中溶出的添加物、单体及其降解物等)进行初步的风险评估并明确潜在的目标浸出物,并依据提取试验研究中获得的可提取物种类和水平信息,建立灵敏的、专属的分析方法,以指导后续的浸出物研究(迁移试验)。
本项目将参考NMPA发布的指导原则如《化学药品注射剂生产所用的塑料组件系统相容性研究技术指南(试行)》、《化学药品与弹性体密封件相容性研究技术指导原则(试行)》和《化学药品注射剂与塑料包装材料相容性研究技术指导原则(试行)》,对氯化钠注射液注射液生产过程中用到的316L不锈钢、玻璃和硅胶进行模拟提取。
4.1 提取条件的选择
参考NMPA发布的《化学药品注射剂生产所用的塑料组件系统相容性研究技术指南(试行)》:开展提取试验时,应对提取方式、溶剂、提取比例、温度、时间等进行合理选择和设计。选择提取溶剂的关键因素包括:根据液体特点,考虑极性、pH、离子强度等,适当对提取溶液进行替换或者调整;提取溶液的用量应保证组件系统表面积与溶液体积比在合适范围内;提取温度和时间通常不低于实际生产过程中组件系统和液体之间的接触温度和时间;提取过程中组件系统的处理方式一般应与实际使用时的处理方式保持一致。
4.1.1、提取溶剂:基于上述指导原则,本项目将选择1)制剂、2)酸性提取溶液(pH=3,pH值应不高于药品的实际处方)、3)碱性提取溶液(pH=10,pH值应不低于药品实际处方)、4)50%乙醇溶液4个提取溶剂,进行提取试验研究。
4.1.2、提取条件:根据本品生产过程中接触最长时间19h、最高接触温度80℃,选择比生产工艺更剧烈的条件,100℃加热24h,进行提取试验研究。
表5:提取条件及检测项目
序号 | 提取溶剂 | 材质 | 提取条件 | 检测项目 | ||
元素检测(ICP-MS) | 挥发物/半挥发物检测(GC-MS) | 不挥发物检测 (LC-MS/MS) | ||||
1 | 制剂 | 316L不锈钢 | 100℃,24h | √ | N.A. | N.A. |
2 | pH3酸性溶液 | √ | N.A. | N.A. | ||
3 | pH10碱性溶液 | √ | N.A. | N.A. | ||
4 | 制剂 | 玻璃 | 100℃,24h | √ | N.A. | N.A. |
5 | pH3酸性溶液 | √ | N.A. | N.A. | ||
6 | pH10碱性溶液 | √ | N.A. | N.A. | ||
7 | 制剂 | 硅胶 | 100℃,24h | √ | √ | √ |
8 | pH3酸性溶液 | √ | √ | √ | ||
9 | pH10碱性溶液 | √ | √ | √ | ||
10 | 50%乙醇溶液 | N.A. | √ | √ | ||
注:“√”为检测项目,“N.A.”为此项目检测不涉及。
4.2 提取样品溶液的制备
模拟溶剂1(制剂):氯化钠注射液注射液。
模拟溶剂2(pH3缓冲液):称取14.9g氯化钾,置1000ml聚四氟乙烯瓶中,用水稀释至刻度,配制成0.2mol/L的氯化钾溶液。用0.2mol/L盐酸调节pH至3±0.1。
模拟溶剂3(pH10缓冲液):称取14.2g磷酸氢二钠,置1000ml聚四氟乙烯瓶中,用水稀释至刻度,并用0.1mol/L的盐酸溶液或氢氧化钠溶液调节pH至10±0.1。
模拟溶剂4(50%乙醇溶液):量取无水乙醇500ml与500ml水,混匀,即得。
316L不锈钢提取液:将316L不锈钢切割成每段长为4cm,清洗干净后滤纸吸干,作为供试品,放入聚四氟乙烯瓶内,分成3组,每组2段(按双面计,总面积约为88cm2),第一组瓶内加入制剂75ml,第二组瓶内加入pH3缓冲液75ml,第三组瓶内加入pH10缓冲液75ml,于100℃加热24h后取出,即得提取样品1、2、3。以相应提取溶剂装入聚四氟乙烯(PTFE)瓶内同法作为空白溶液。
玻璃提取液:将玻璃切割成每段长为4cm,清洗干净后滤纸吸干,作为供试品,放入聚四氟乙烯瓶内,分成3组,每组1段(按双面计,总面积约为113cm2),第一组瓶内加入制剂95ml,第二组瓶内加入pH3缓冲液95ml,第三组瓶内加入pH10缓冲液95ml,于100℃加热24h后取出,即得提取样品4、5、6。以相应提取溶剂装入聚四氟乙烯(PTFE)瓶内同法作为空白溶液。
硅胶提取液:将硅胶管、硅胶垫清洗干净,滤纸吸干,硅胶管切成每段长为2cm,每个硅胶垫按通过圆心的两条垂直线剪成4块,每块再剪成4段(每段约1cm),作为供试品,放入聚四氟乙烯瓶内,分成4组,每组由两段硅胶管和一块硅胶垫组成(硅胶垫按双面计,硅胶管按内表面和外表面计,总面积为24cm2),第一组瓶内加入制剂30ml,第二组瓶内加入pH3缓冲液30ml,第三组瓶内加入pH10缓冲液30ml,第四组瓶内加入50%乙醇溶液30ml,于100℃加热24h后取出,即得提取样品7、8、9、10。以相应提取溶剂装入聚四氟乙烯(PTFE)瓶内同法作为空白溶液。
表6:供试品表面积
氯化钠注射液注射液-工艺组件相容性研究模拟提取试验 | |||||||
名称 | 外直径/cm | 内直径/cm | 每段长 | 外表面积/cm2 | 内表面积/cm2 | 每段面积/cm2 | 每组面积/cm2 |
316L不锈钢 | 1.9 | 1.6 | 4cm | 23.8761 | 20.1062 | 44.0 | 88 |
玻璃 | 4.7 | 4.3 | 4cm | 59.0619 | 54.0354 | 113.1 | 113 |
硅胶管 | 1 | 0.5 | 2cm | 6.2832 | 3.1416 | 9.4 | 24 |
硅胶垫 | 5.1 | 3.5 | 4块 | 20.4282 | 9.6211 | 5.4 | |
表7:供试品表面积与提取溶剂的比例
组件材质 | 提取比例(cm2/ml) | 限度浓度(μg/ml) | 备注 |
316L不锈钢 | 1.2 | N.A. | 供试品厚度>1mm |
玻璃 | 1.2 | N.A. | |
硅胶 | 0.8 | 0.72 | |
计算公式:限度浓度(μg/ml)=AET(μg/cm2)×提取比例(cm2/ml) | |||
4.3 提取样品的检测
4.3.1元素检测
采用电感耦合等离子体质谱仪(ICP-MS),按表8拟定的项目,对不同工艺组件的3种提取样品溶液(不含有机溶液)中可能存在的元素进行定性全扫描(共73种)。
4.3.1.1所用仪器参数
表8:仪器及参数信息
仪器 | ICPMS 电感耦合等离子体质谱仪(赛默飞iCAP RQplus ICP-MS) | ||
射频功率 | 1200W | 采样深度 | 5.0mm |
等离子体气流量 | 9.0L/min | 辅助气流量 | 0.9L/min |
载气流量 | 0.70L/min | 池气体 | 6.0L/min |
注:仪器参数最终以实验结果进行适当调整为准。
根据元素全扫描结果,综合相关指导原则,及考虑各元素在自然界的丰度、工艺组件中存在的可能性,采用标准工作曲线法,对提取溶液中超出仪器检出限的元素进行半定量检测。
4.3.1.2元素半定量检测
根据氯化钠注射液注射液每日最大用药量所对应的工艺组件接触面积,将各元素的PDE换算成对应供试品溶液的浓度值,设计相应的线性曲线,对提取溶液中超出仪器检测限的元素进行半定量检测。扫描供试品溶液配制、元素限度浓度换算公式设计如下:
稀释剂:量取硝酸7.5ml,用去离子水稀释至1000ml,混匀,即得。
扫描供试品溶液配制:分别量取提取溶液各2.5ml,置不同25ml量瓶,用稀释剂稀释至刻度,摇匀,即得。
将元素PDE换算为供试品溶液中元素限度浓度的公式如下:
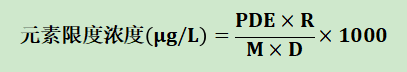
式中:
PDE:元素杂质的每日允许暴露量,μg/天 ;
R:提取比例,cm2/ml;
M:每日最大用药量所对应的工艺组件接触面积,cm2/天;
D:前处理稀释倍数;
1000:L转换成ml的换算系数。
4.3.2挥发性/半挥发性物质检测(硅胶)
挥发性/半挥发物检测涵盖抗氧剂、增塑剂、环硅氧烷类等物质,使用气相色谱-质谱联用仪(GC-MS)完成检测。
量取各提取样品溶液适量(不含有机相),加入二氯甲烷,萃取,静置分层,取二氯甲烷层进行检测,结果将参考相关数据库进行分析。
4.3.3不半挥发性物质检测(硅胶)
不挥发物检测涵盖抗氧剂及降解产物、酯类、硫化物等,使用液相色谱-质谱联用仪(LC-MS/MS)完成检测。
量取各提取样品溶液适量进行检测,结果将参考相关数据库进行分析。
五、提取试验研究结果评估
5.1 元素杂质
将分析测试得到的各元素的浓度值,与计算的元素限度值比较,低于30%元素限度值的,则无需关注;高于30%元素限度值的元素,则需要在浸出物研究中进行检测。此规则不适用于注射途径需评估的元素及制剂中本身含有的元素。
5.2 有机杂质
将分析测试得到的可提取物的浓度值,与其分析评价阈值(AET)进行比较,低于AET时,无论是否具有致癌性,均可忽略其安全风险;高于AET时,则需要在浸出物研究中进行检测。
5.3 目标浸出物的确定
根据提取溶液中提取物的水平、可提取毒性评估结果,结合工艺组件材料配方,确定目标有机浸出物。对于浓度高于AET的元素杂质和有机杂质,可根据其积累水平、潜在安全风险、相关法律法规规定,并结合工艺组件配方选择有代表性的添加剂或者降解产物作为潜在目标浸出物,进行方法学研究,并在迁移试验中关注。
表9-1:目标元素确认依据
序号 | 来源 | 判断依据 |
1 | 提取样品溶液中浓度高于30%元素限度值的元素 | 将根据提取样品溶液的检测结果确定 |
2 | ICH Q3D指导原则中注射剂需要风险评估的元素,共10种 | Cd、Pb、As、Hg、Co、V、Ni、Li、Sb、Cu |
3 | 工艺组件中特意添加的元素 | 请客户提供工艺组件的证明资料及检测报告 |
表9-2:目标有机浸出物确定依据
序号 | 来源 | 判断依据 |
1 | 提取样品溶液中浓度高于AET的化合物 | 将根据提取样品溶液的检测结果确定 |
2 | 具有潜在安全风险的物质 | 将根据提取样品溶液的检测结果确定 |
3 | 硅胶中特意添加的物质及单体 | 请客户提供硅胶的证明资料及检测报告 |
4 | 硅胶中常用的添加剂如:抗氧剂及降解产物、增塑剂等 | 根据提取样品溶液的检测结果并结合硅胶的成分信息确定 |
六、方法学验证
6.1 元素杂质
参考《中国药典》四部通则9101、ICH Q2、USP<232>、USP<233>对目标元素进行方法学验证,验证参数主要包括:系统适用性、专属性、线性及范围、检测限、定量限、准确度、精密度(重复性和中间精密度)、溶液稳定性等。具体操作与可接受标准如下表所示。
表10:元素方法验证及可接受标准
验证项目 | 验证方法 | 可接受标准 | |
系统适用性 | 同一份的对照品溶液(100%限度水平)连续进样6次,用6次的测定结果进行评价。 | RSD≤20%。 | |
专属性 | 1.空白溶液1份; 2.对照品溶液1份(100%加标水平); 3.样品空白溶液1份; 4.供试品溶液1份; 5.供试品加标溶液1份(100%加标水平); 5份溶液各进样一次。 | 空白溶液和样品空白溶液对各元素杂质测定无干扰;各元素杂质供试品加标溶液测得浓度减去供试品溶液测得浓度的值在对照品溶液测得浓度的70%~150%范围内。 | |
线性及范围 | 配制线性溶液,上机检测,绘制线性曲线。 | 各元素线性方程的相关系数r>0.990。 | |
检测限 | 7份空白样品溶液连续进样,用3倍空白溶液响应值的标准偏差作为理论检测限。 | 理论检测限≤实际检测限(10%元素限度值)。 | |
7份空白样品溶液连续进样,用10倍空白溶液响应值的标准偏差作为理论定量限。 | 理论定量限<实际定量限(30%元素限度值)。 | ||
6份供试品加标溶液(30%限度水平)。 | 各元素测得浓度RSD≤20%。 | ||
准确度 | 1.配制3份相当于30%限度水平的供试品加标溶液; 2.配制3份相当于100%限度水平的供试品加标溶液; 3.配制3份相当于150%限度水平的供试品加标溶液。 | 回收率70%~150%,回收率RSD≤25%。 | |
精密度 | 重复性 | 配制6份供试品加标溶液(100%限度水平) | 各元素杂质的回收率应在70%~150%之间,回收率的RSD≤20%(n=6)。 |
中间精密度 | 由另一名试验人员独立同法配制6份供试品加标溶液(100%限度水平),各进样1次。 | 各元素杂质的回收率应在70%~150%之间,回收率的RSD≤20%(n=6);RSD≤25%(n=12)。 | |
溶液稳定性 | 取供试品溶液、对照品溶液和供试品加标溶液室温放置0、1、2小时后,进行检测。(注:供试品溶液中各元素杂质的测得浓度均低于检测限,可直接判定供试品溶液稳定) | 供试品溶液、对照品溶液、供试品加标溶液各时间点检测结果与0小时比较,各元素杂质的浓度变化值在20%范围内。 | |
6.2 有机杂质
参考《中国药典》2020年版四部通则9101、《化学药物质量控制分析方法验证技术指导原则》、ICH Q2等对目标有机浸出物进行方法学验证验证参数主要包括:专属性、线性及范围、检测限、定量限、准确度、精密度(重复性和中间精密度)、耐用性、溶液稳定性。具体操作与可接受标准如下表所示。
表11:有机物方法验证及可接受标准
验证项目 | 验证方法 | 可接受标准 |
系统适用性 | 1.对照品溶液1连续进样5次; 2.对照品溶液2连续进样2次。 | 连续进5针对照品溶液所得各杂质峰面积的RSD不得过10.0%; 保留时间RSD不得过1.0%; 7针对照品溶液各杂质f值的RSD不得过10.0%。 |
专属性 | 1.配制空白溶液1份; 2.配制对照品溶液1份; 3.供试品溶液1份; 4.供试品加标溶液1份(100%加标水平); 4份溶液各进样一次。 | 空白溶液对目标物检测应无干扰; 供试品溶液中其他成分对目标物检测应无干扰; 供试品加标溶液中目标物与对照品溶液中目标物出峰时间一致; 目标峰与相邻峰的分离度符合要求。 |
检测限 | 检测限溶液,进样1次。 | 目标峰信噪比≥3。 |
定量限 | 定量限溶液,进样6次。 | 目标峰信噪比≥10; 6次进样峰面积RSD应不得过15.0%。 |
线性和范围 | 每个线性溶液进样1次。 | 线性相关系数的r>0.990。 |
准确度 | 1.配制3份相当于30%限度水平的加标供试品溶液; 2.配制3份相当于100%限度水平的加标供试品溶液; 3. 配制3份相当于150%限度水平的加标供试品溶液。 | 回收率70%~125%,回收率RSD≤20%。 |
精密度(重复性) | 配制6份供试品加标溶液(100%限度水平) | 6份重复性溶液中各目标峰的回收率应在70%~125%之间,回收率RSD应不得过15.0%。 |
精密度(中间精密度) | 由另一名试验人员独立同法配制对照品溶液1、对照品溶液2、2份供试品溶液、6份供试品加标溶液(100%限度水平),按系统适用性及重复性验证要求验证。 | 6份中间精密度溶液中各目标物的回收率应在70%~125%之间,回收率的RSD应不得过15.0%。两名试验人员所得12份供试品加标溶液中各目标物回收率RSD应不得过20.0%。 |
溶液稳定性 | 1.对照品溶液; 2.供试品加标溶液。室温放置,于不同时间点取样检测。 | 对照品溶液与供试品加标溶液室温放置,于不同时间点取样检测,各时间点检测结果与0小时比较,各目标物峰面积的比值为70%~125%。 |
耐用性 | 改变进样口温度(±5℃)、流速(±0.1ml/min)、分流比(±1)等条件。 | 系统适用性均符合要求,各目标物的回收率的应在70%~125%,回收率RSD值不得过20.0%。 |
七、浸出物研究
浸出物研究将根据实际生产过程中工艺组件和药品的接触情况,考察实际生产工艺中可能出现在药品中的浸出物信息。
研究样品优先选择由委托方提供的3批工艺液体进行测试,若实际条件无法实现,也可以提供0点或其他时间点的稳定性样品。
采用经过验证后的方法对以上样品进行元素和有机化合物的含量测试。
八、相容性研究结论
结合上述结果、评价,确认本品是否与工艺组件相容。
免责声明:本站提供的一切文章和内容信息仅限用于学习和研究目的;不得将上述内容用于商业或者非法用途,否则,一切后果请用户自负。本站信息来自网络收集整理,版权争议与本站无关。我们非常重视版权问题,如有侵权请邮件与我们联系处理。敬请谅解!
本文最后更新于2023-11-30 16:17:32,如果你的问题还没有解决,可以加入交流群和群友们一起讨论。


 在线客服
在线客服
 快速发布
快速发布
 文章查询
文章查询
 分析化学
分析化学
 我的消息
我的消息
 文章发表
文章发表

 回到顶部
回到顶部