
分析方法在药学质量研究中扮演重要的角色,普析手段目前还是在分析领域占据重要的位置,一般尤以HPLC-UV占比最大,一个好的分析方法能有效检出样品中存在的所有杂质,不遗漏某些杂质的检出,以免误导工艺人员正确的开发。
分析方法的建立往往在原料药的质量研究中应用颇多,选择合适的分析方法建立思路,快速过渡,以QBD思想快速筛选,以获得最优的分析方法,对于质量研究尤为重要。以下是我在实际工作应用中对于方法建立等方面的一些心得。
分析方法建立的常规思路
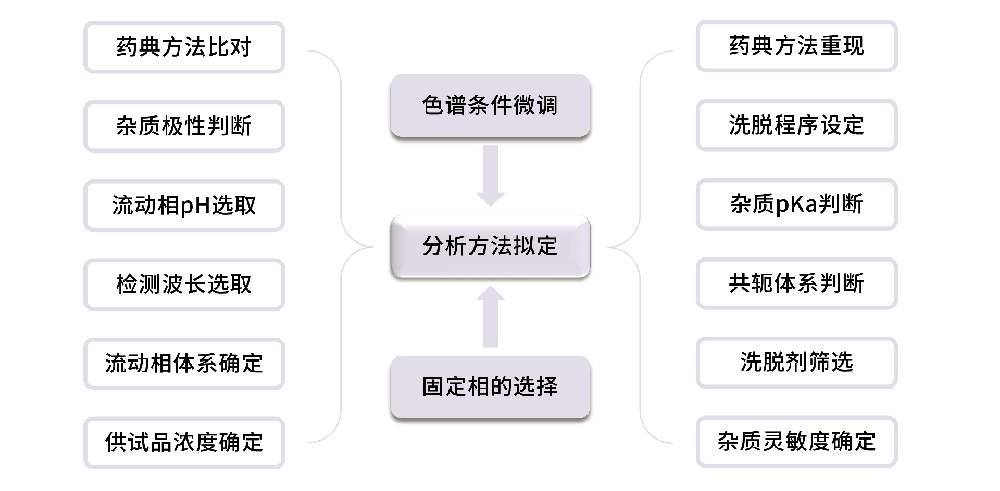
仿制药分析方法建立,一般有大量参考文献,可以在文献基础上进行优化,事半功倍!
创新药分析方法建立,一般参考文献较少,在理论与经验结合的基础上快速建立方法!在HPLC-UV建立之初,我们通常需要完成以下准备:
1、原料药与杂质结构分析,获得重要信息
原料药与杂质pKa判断,以选取流动相体系最佳pH值。

流动相pH值一般选取pKa±2范围之外,以保障化合物处于分子状态,减少电离,此状态下化合物疏水性较强,色谱柱保留时间较长,能获得良好的峰型。
酸性化合物,可以选择非缓冲的流动相进行pH调节,用单一的酸溶液(一般常用磷酸)进行色谱分离,酸的浓度应足以使pH值比需要值低的多。
碱性化合物,选择是比较有限的,TEA/氨水可以用于调节到高的pH值,但是要求色谱柱能够耐碱比较好。
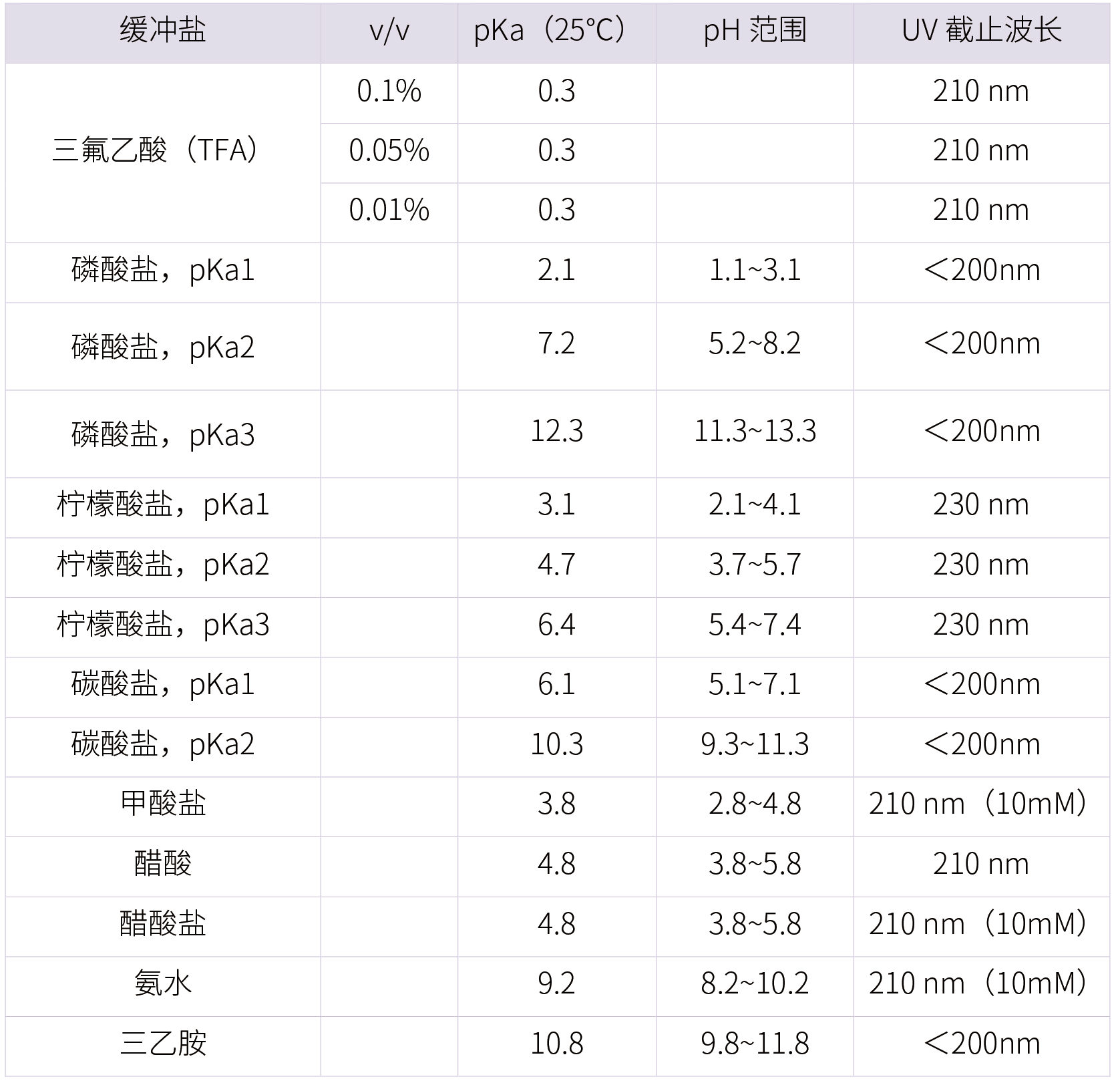
2、流动相缓冲盐选取
缓冲体系选择的要求:能适当控制离子化、与检测器兼容性、缓冲盐溶液与有机相的互溶性、流动相的pH是否在色谱柱的安全范围内。
常用的缓冲体系的盐一般为磷酸盐与醋酸盐体系,其pKa见下表:
3、洗脱剂的选取
反相体系下,常用的洗脱剂为甲醇、乙腈与四氢呋喃。三种溶剂对比如下:

3、洗脱剂的选取
a. 影响分离度的总因素

选择性:α;柱效:N;保留因子:N
选择性是影响分离度的最关键因素 :
改变固定相
改变流动相
柱效是最容易预测和改变的因素; 最理想的保留范围:1<K<10 5 方法建立过程中其他的关注点 a. API与杂质溶解性 直接影响:杂质提取不充分,检测结果不平行,重现性结果差! 间接影响:回收率波动较大,方法建立与方法验证结果不匹配。 主成分在稀释剂中的饱和溶解度,最好满足饱和溶解度大于3倍的供试品浓度。 b. 灵敏度 检测灵敏度不满足,高浓度溶解性会有问题,浓度太大,峰拖尾影响检测,易损坏色谱柱,在遗传毒性杂质方法建立过程中尤为突出。 c. 酮式烯醇式互变 同一化合物有两个响应接近的峰,主峰伴随着一个较大的未知峰,峰有台阶,互变影响峰型。 具体可参考占小兵老师的“HPLC方法开发需注意的典型特殊结构化合物专题:酮式烯醇式互变”。 d. 溶剂效应
b. 影响分离度的色谱条件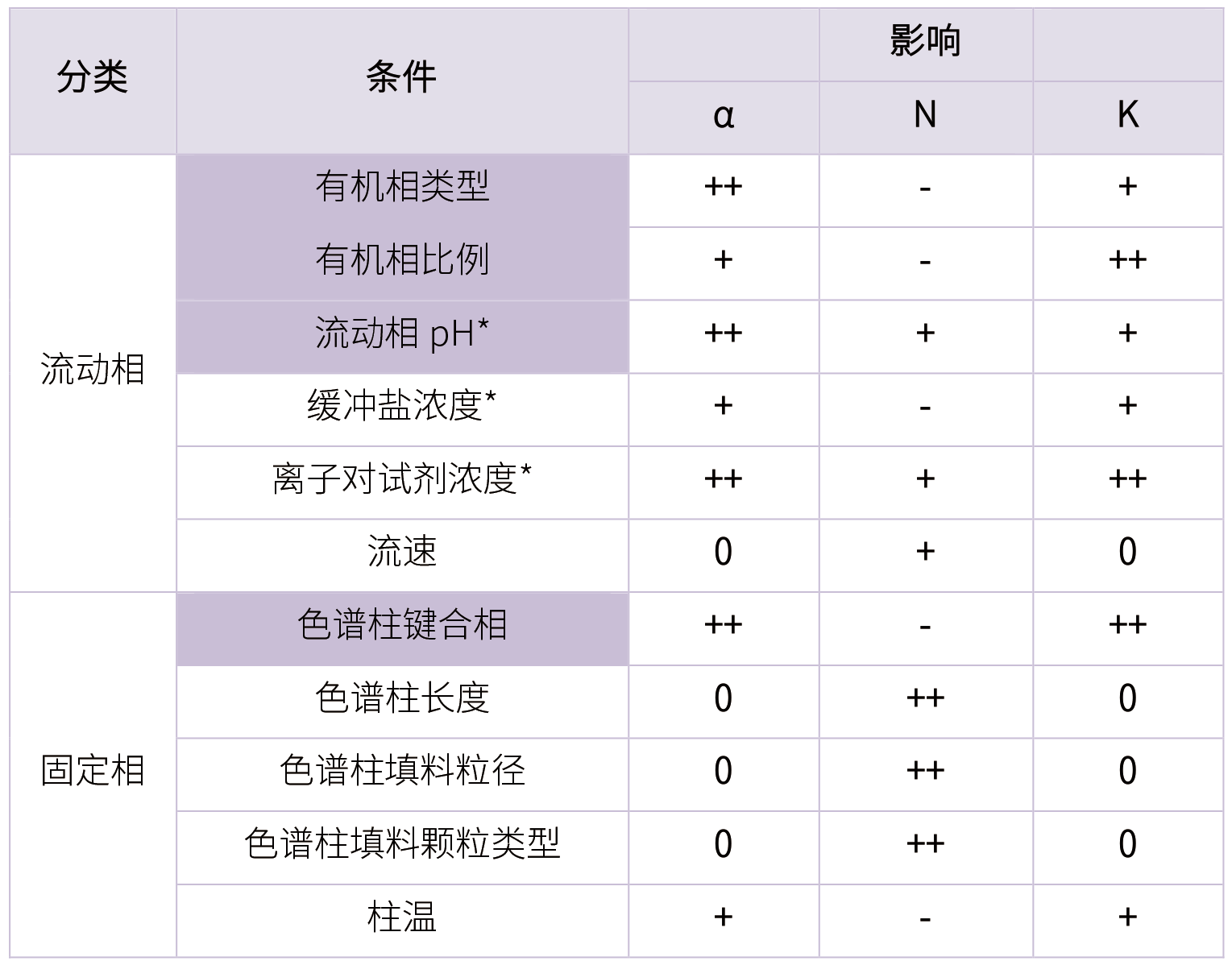
优先采用流动相体系,降低溶剂强度(如增加水相比例,调整pH等)。
助溶剂选择,最好控制在5%以内,避免强溶剂效应。 e. 极性化合物弱保留
强保留型色谱柱(AQ色谱柱…..),离子交换色谱柱,离子对试剂, HILIC模式。
离子对试剂
酸性化合物:四丁基氢氧化铵、四丁基氯化铵、四丁基溴化铵、四丁基硫酸氢铵等。
碱性化合物:烷基磺酸盐类,如戊烷磺酸钠、辛烷磺酸钠、庚烷磺酸钠等。三氟乙酸、五氟丙酸等全氟取代的直连有机酸等。
防止漏检杂质,最重要的就是需要进行分析方法可靠性评估,以判断分析方法的适用性。 分析方法的建立,是一个系统的工程,在有参考文献的条件下,需要评估方法的可靠性;线性洗脱程序的合理性,也是检验方法适用性重要的一环。
分析方法可靠性评估
常规做法:
根据合成工艺路线及降解途径分析可能产生杂质的全面性。
采用强制降解试验来判断方法的适用性。
延长梯度洗脱高比例洗脱剂洗脱时间,防止小极性杂质漏检。
药典方法不一定是最好的! 1.在有参考标准前提下,要理性判断分析方法的可靠性,以免获得假阴性结果误导工艺开发,要时刻谨记:药典方法不一定是最好的!
方法对比如下: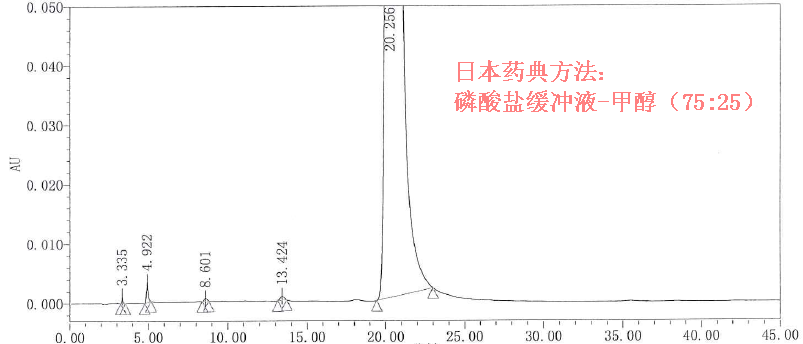
免责声明:本站提供的一切文章和内容信息仅限用于学习和研究目的;不得将上述内容用于商业或者非法用途,否则,一切后果请用户自负。本站信息来自网络收集整理,版权争议与本站无关。我们非常重视版权问题,如有侵权请邮件与我们联系处理。敬请谅解!
本文最后更新于2022-11-28 22:18:48,如果你的问题还没有解决,可以加入交流群和群友们一起讨论。


 在线客服
在线客服
 快速发布
快速发布
 文章查询
文章查询
 分析化学
分析化学
 我的消息
我的消息
 文章发表
文章发表

 回到顶部
回到顶部