

一、标准的修订原则《食品安全国家标准 预包装食品标签通则》(GB 7718-2011)自颁布实施以来,对保障食品安全、规范食品市场、引导科学消费发挥了重要作用。随着食品产业发展,预包装食品市场不断丰富,标签形式多种多样,GB 7718-2011需要修订以适应食品标签的管理需求。标准修订遵循《中华人民共和国食品安全法》的规定,
注射剂包装系统密封性符合要求,通常是指包装系统已经通过或能够通过微生物挑战测试。广泛意义指不存在任何影响药品质量的泄漏。应确定最大允许泄漏限度。密封性检查方法的开发和验证,关注方法选择及灵敏度,方法需进行合理验证;稳定性初期和末期外其他时间点可采用包装系统密封性测试作为无菌检查的替代;注射剂包装系统
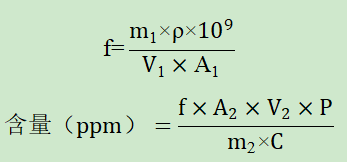
一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测注射剂中N-亚硝基哌啶的含量。参考注射剂说明书,其规格为75mg,每日最大用量为150mg。根据EMA人用药品中的亚硝胺杂质上市许可人问答中N-亚硝基哌啶的PDE=1300ng/day,计算N-亚硝基哌啶的限度为1300μg/150mg= 8.7ppm。根据N-亚硝基哌啶
一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测注射剂中N-亚硝基哌啶的含量。参考注射剂说明书,其规格为75mg,每日最大用量为150mg。根据EMA人用药品中的亚硝胺杂质上市许可人问答中N-亚硝基哌啶的PDE=1300ng/day,计算N-亚硝基哌啶的限度为1300μg/150mg= 8.7ppm。根据N-亚硝基哌啶
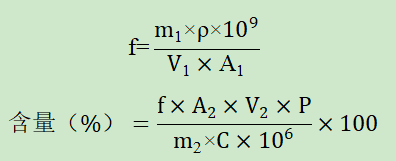
一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测注射用中基因毒性杂质氯甲烷、氯乙烷和2-氯丙烷的含量。参考注射用说明书,其规格为75mg,每日最大用量为150mg。根据ICH M7,氯甲烷和氯乙烷的终身AI值分别为1361μg/天、1810μg/天,经计算氯甲烷、氯乙烷的限度分别为1361μg/150mg=0

一、研究目的化学药品生产过程中直接接触的工艺组件,可能与液体接触并发生相互作用,导致相关浸出物的产生和积累。浸出物在液体中持续存在并最终传递至终产品中,可能影响产品质量和/或患者安全。本方案参考《化学药品注射剂生产所用的塑料组件系统相容性研究技术指南(试行)》、《化学药品与弹性体密封件相容性研究技术指

一、验证目的本试验的目的是为了有效的检测氟雷拉纳中4-甲酰-2-甲基苯甲酸甲酯和3,5-二氯溴苯的含量。产品中4-甲酰-2-甲基苯甲酸甲酯和3,5-二氯溴苯限度均为1.5ppm。根据此限度要求,对氟雷拉纳中4-甲酰-2-甲基苯甲酸甲酯和3,5-二氯溴苯的检测方法进行验证,验证内容包括系统适用性、专属性、检测限、定量限、线性及范围、准

一、背景介绍给药器具作为一种医疗器械,用于将药物不经过任何生物屏障直接输注进入人体循环系统的一种独特给药器械。由于给药过程中,给药器具会和药液短时的接触,在临床使用过程中可能会对药液的活性成分或者功能性辅料产生吸附作用,尤其对于一些低剂量给药的药物,吸附作用对药物的治疗效果影响更大,如一次性无菌注射
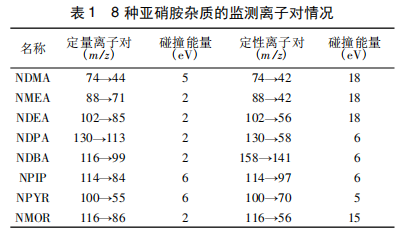
头孢菌素类药物是以天然头孢菌素 C作为原料,经过半合成改造其侧链而得到的一类抗菌药物,也是目前临床应用最广泛的一类 抗菌药物。头孢孟多酯钠和盐酸头孢替安属于第二 代头孢菌素类药物,对革兰阴性杆菌所产生的的 β- 内酰胺酶的稳定性较第一代头孢菌素强,临床上用 于呼吸系统、皮肤和软组

概述影响药物稳定性的因素主要包括三个方面:化学不稳定、物理不稳定、生物不稳定。(1)化学不稳定是指药物由于氧化、水解、聚合、异构、脱羧,以及药物相互作用而产生的化学反应。(2)物理不稳定要是由于温度、湿度等因素引起物理状态的改变(如无定型、多晶型)。(3)生物不稳定是指微生物污染、发酵、分解、变质

我们做有机合成的会接触到很多参数,比如展开剂极性大小,各种溶剂的参数,包括毒性,沸点,溶解度等等。常用溶剂互溶表,压力单位换算表,常用显色剂配置和使用方法,以及各种残留溶剂的核磁。我们就不一一赘述了。这些参数记忆起来还是比较有困难的,最好的方法就是能够把这些参数能汇总成表格

目前药行的小伙伴们接触较多的剂型是以片剂、胶囊剂、针剂居多。但是,对于像糖浆剂、贴膜剂,喷雾剂,软膏剂、栓剂却不是很常见。今天我们就换换方向,聊一聊大家见得不多的剂型——软胶囊(胶丸)。随着软胶囊应用越来越广泛,软胶囊开始被争相研发、仿制。软胶囊参比制剂的逆向解析工作也就成为了软胶囊研发至关重
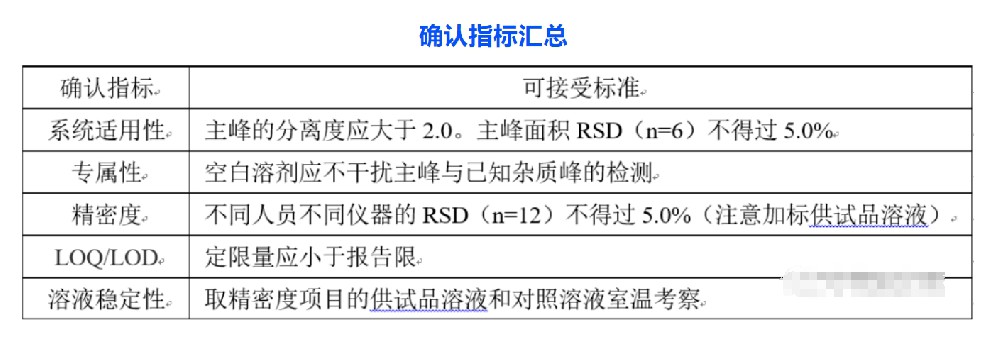
制剂通常的发补内容格式:参照各国药典,结合所用原料药的合成路线,对可能存在的杂质进行全面分析,考察自拟有关物质检查方法对各已知杂质的分离和检出能力,必要时对有关物质方法进行优化,提供自拟方法的验证资料。尬点:原料药采用自拟方法(申报时药典没收载)虽然重新全面考察了已知杂质的分离度和检出限均满足要求,

一、摘要本试验对XXXXXX有限公司提供的聚丙烯输液袋(含聚丙烯组合盖)进行相容性研究。参考《化学药品注射剂与塑料包装材料相容性研究技术指导原则(试行)》,选择合适的模拟溶剂(制剂溶液、pH3酸性提取液、pH10碱性提取液、15%乙醇溶液)和提取条件对聚丙烯输液袋(含聚丙烯组合盖)进行模拟提取试验,预测可能迁移到药

1、强制试验强力试验Stress Testing,又称为强力破坏试验,或强制降解试验,为原料药和产品设计符合监管要求的稳定性计划的重要步骤,该类试验有助于确定可能的降解产物,而这些降解产物又可帮助了解降解途径和分子内在的稳定性,并论证使用的分析方法是否能反映产品的稳定性。强制破坏试验的类型将取决于各种原料药的性质及
1、中国专利公布公告网网址:http://epub.sipo.gov.cn/简介:网站包括自1985年9月10日以来公布公告的全部中国专利信息,其检索功能可以按照发明公布、发明授权、实用新型和外观设计四种公布公告数据进行查询,数据主要包括中国专利公布公告信息,以及实质审查生效、专利权终止、专利权转移、著录事项变更等事务数据信息。2、

打开浏览器,输入“FDA”进入或打开链接https://www.fda.gov/,进入FDA主页,如“图1”图1点击“图1”中“Drugs”进入药物检索页,网址:https://www.fda.gov/drugs,一般情况从“图2”中“Drug Approvals and Databases”可获取绝大多数FDA公开的原研药品信息。图2点击“图2”中的“Drug Approvals and Databases”进入药物
一提起药品说明书,大家肯定会说它是用来指导患者安全用药的东西,和药品研发似乎关系不大。当我们不知道药品的用法用量,才会看看说明书上是怎么写的。其实,大家对于药品说明书的了解不应该仅此而已。对于药研人来说,作为药品的“身份证”,药品说明书提供了很多药品安全性、有效性的重要科
根据药品注册的国际技术要求(ICH)中杂质的含义, 杂质分为有机杂质、无机杂质和残留溶剂。有关物质是杂质的一种, 主要是指有机杂质。它的检查方法很多, 而其中的HPLC法具有专属性强、分离效果好、灵敏度高、分析速度快、重复性好、操作简便等诸多优点, 且能与多种类型检测器联合应用, 能满足大多数化合物种类的检测需要, 比较
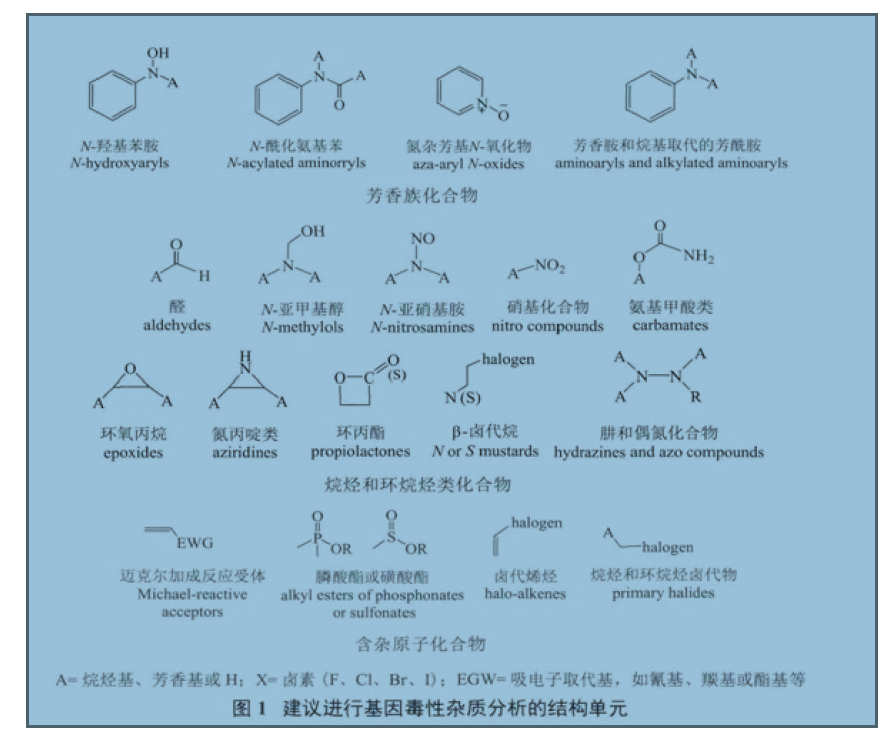
1 从宏观上解读杂质1.1 杂质与药物不良反应的关系 很多同仁都认为杂质与药物的不良反应息息相关,认为杂质越小或越少、临床不良反应发生几率也就越小或越少,进而在进行杂质研究与控制时,力求面面俱到、尽善尽美。殊不知,引起药物不良反应的原因是多方面的,并不仅仅是药物中的杂质。人用药品注册技术要求国
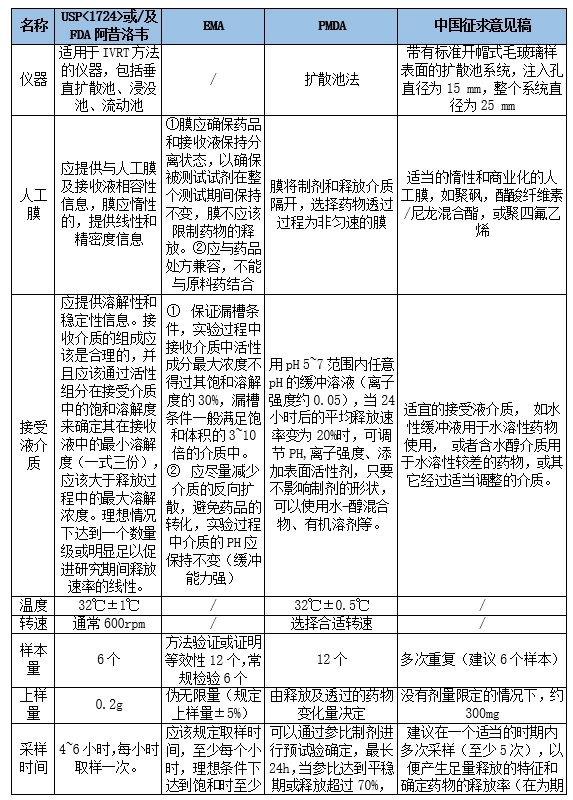
体外释放试验(in vitro release test,IVRT)是表征和评价半固体制剂性能的有效手段,体外释放速率可以反映药物的溶解度、粒径、剂型流变性等多种理化参数的综合作用,可辨别处方和工艺变化对制剂的影响,是产品开发、质量控制、稳定性考察及产品批准后变更的重要质量控制项目。&nbs
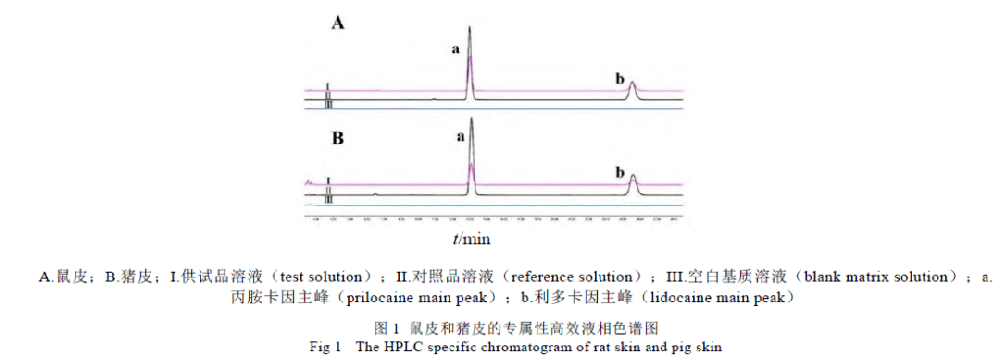
目的:综合国内外关于半固体制剂的体外药物释放试验的研究,探讨关于体外药物释放试验各装置特点及试验条件的选择并判断释药模型。方法:根据国内外31篇文献,对如何根据所研究半固体制剂的特点选择合适装置及试验条件进行体外药物释放试验进行综述。结果:通过药物的体外释放结果判断出药物符合的释药模型并阐明释药机理。
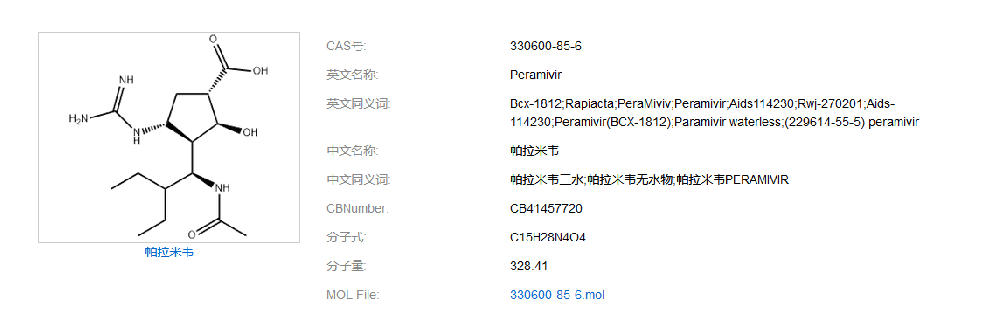
如何建立帕拉米韦质量标准,现在百度检索:同时在药物在线(https://www.drugfuture.com/standard/):各国药典均未收录载帕拉米韦。无药典收载、无进口标准:医学百科(https://www.yixue.com):帕拉米韦水合物是美国BioCryst 公司开发的以流感病毒表面糖蛋白神经氨酸酶为作用靶点的新型环戊烷类抗流感病毒制剂,是世界首
亚硝胺英文来源药物名称AI值(ng/day)CPCA潜在分类发表日期N-Methyl-N-nitrosophenethylamine(13256-11-6, NMPEA)-N-甲基-N-亚硝基甲胺85-Jan-233-((Ethyl(nitroso)amino)methyl) benzenesulfonateDextromethorphan右美沙芬18129-Jul-24N-Nitroso-2,4-thiazole amine /N-((2-isopropylthiazol-4-yl)methyl)-N-methylnitrous

1 国内外遗传毒性杂质监管现状1 从宏观上解读杂质1.1 杂质与药物不良反应的关系很多同仁都认为杂质与药物的不良反应息息相关,认为杂质越小或越少、临床不良反应发生几率也就越小或越少,进而在进行杂质研究与控制时,力求面面俱到、尽善尽美。殊不知,引起药物不良反应的原因是多方面的,并不仅仅是药物中的杂质。人用药品

一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测盐酸度洛西汀中基因毒性杂质N-亚硝基二甲胺(NDMA)和1-氟萘的含量。根据客户要求,N-亚硝基二甲胺(NDMA)限度为0.8ppm,1-氟萘限度为12.5ppm。根据此限度要求,对盐酸度洛西汀中NDMA和1-氟萘的检测方法进行验证,验证内容包括系统适用
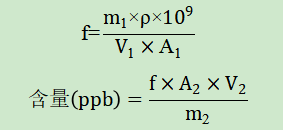
一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测头孢地尼中N-亚硝基二甲胺(NDMA)和N-亚硝基二乙胺(NDEA)的含量。参考头孢地尼胶囊的说明书,每日最大用量为0.3g,根据FDA发布的《人用药中亚硝胺杂质控制》行业指南,NDMA和NDEA每日可接受摄入量分别为:96.0ng/天和26.5ng/天,计算



 在线客服
在线客服
 快速发布
快速发布
 文章查询
文章查询
 分析化学
分析化学
 我的消息
我的消息
 文章发表
文章发表

 回到顶部
回到顶部