

注射剂包装系统密封性符合要求,通常是指包装系统已经通过或能够通过微生物挑战测试。广泛意义指不存在任何影响药品质量的泄漏。应确定最大允许泄漏限度。密封性检查方法的开发和验证,关注方法选择及灵敏度,方法需进行合理验证;稳定性初期和末期外其他时间点可采用包装系统密封性测试作为无菌检查的替代;注射剂包装系统
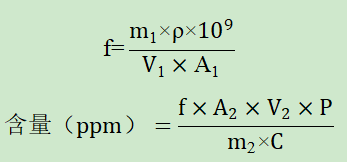
一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测注射剂中N-亚硝基哌啶的含量。参考注射剂说明书,其规格为75mg,每日最大用量为150mg。根据EMA人用药品中的亚硝胺杂质上市许可人问答中N-亚硝基哌啶的PDE=1300ng/day,计算N-亚硝基哌啶的限度为1300μg/150mg= 8.7ppm。根据N-亚硝基哌啶
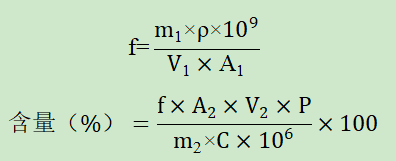
一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测注射用中基因毒性杂质氯甲烷、氯乙烷和2-氯丙烷的含量。参考注射用说明书,其规格为75mg,每日最大用量为150mg。根据ICH M7,氯甲烷和氯乙烷的终身AI值分别为1361μg/天、1810μg/天,经计算氯甲烷、氯乙烷的限度分别为1361μg/150mg=0
有关物质泛指在药品的生产与储存过程中产生的工艺杂质或降解产物。一个经过严格验证的方法才能更有效地控制产品的纯度,进而降低毒副作用的产生。我们对有关物质的控制方法一般为面积归一化法、加校正因子的自身对照法和外标法。《中国药典》2020年版,在《药品质量标准分析方法验证指导原则》中新增了对校正因子的验
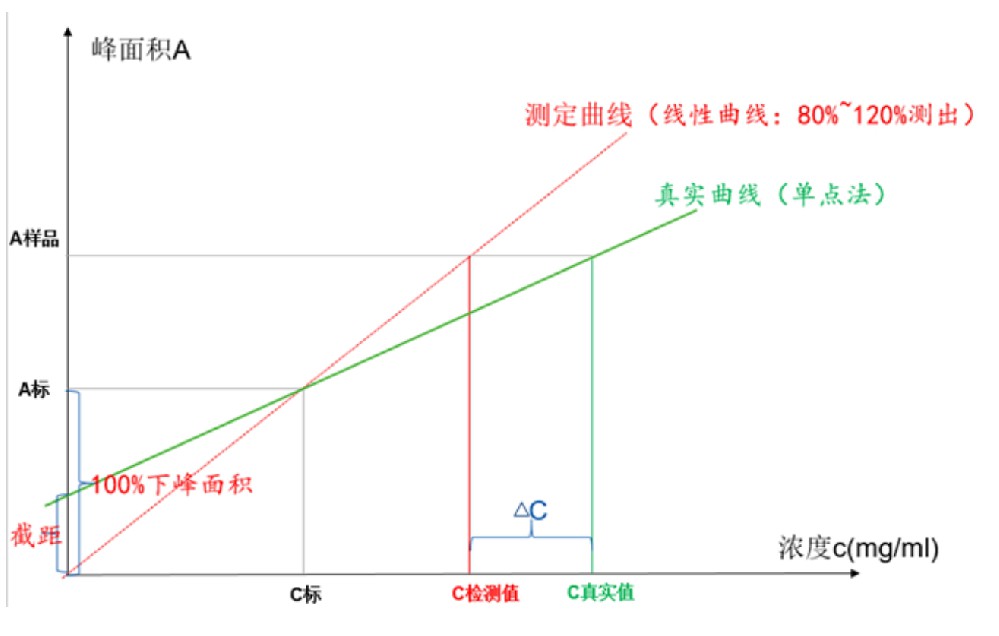
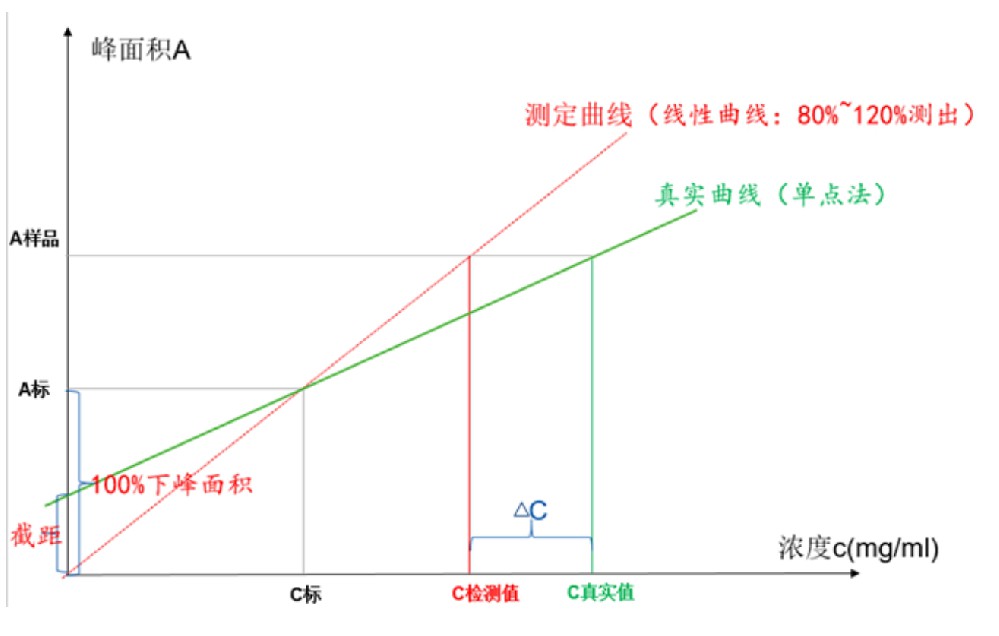
关于分析方法验证线性考察是定量分析的基础和必要条件,定量是基于呈一元一次方程的线性关系进行的,除了需要考察相关系数以确定是否成线性,还必须考察回归方程的截距问题,通常使用两种方法,一种为Y轴截距绝对值与标准(100%)之比法,另一种为剩余标准差标准(100%)之比法,二种方法均能反应截距的大小,对于含量应该不

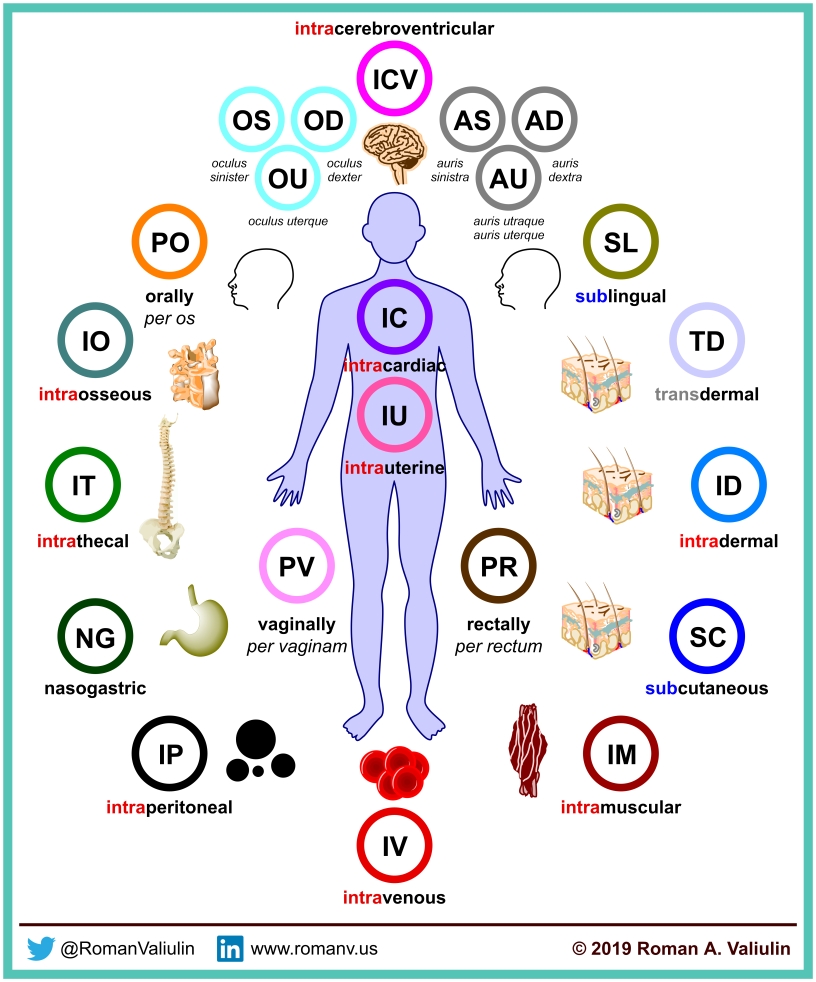
给实验动物用药需要仔细考虑和计划,以优化给动物的药剂递送,同时最大限度地减少手术中的潜在不良反应。对于所有物种,都有许多不同的物质给药途径。必须考虑给药量、给药部位、物质的pH值和其他因素,以完善该技术。在研究的这一方面,训练不足或不注意细节可能会对实验动物产生无意的不良影响,并导致结果混乱。给实验动
仿制药是指与原研药剂量、安全性和效力(strength)(不管如何服用)、质量、作用(performance)以及适应症(intended use)上相同的一种仿制品。原研药专利到期以后,其他国家和制药厂即可生产仿制药。那么作为药研人员应该根据仿制的对象,搜集整理那些信息内容呢?又该怎么查询这些信息呢?

化学分析网记得在学校的时候,一般合成的化合物,先点板(TLC)监测反应,有产物生成后,再过柱子,打核磁,质谱会最后做。随着技术的不断发展和高校科研条件的不断改善,越来越多的科研机构和制药公司都配备了LCMS,既可以用来监测反应,又可以用来表征产物的纯度。小编发现,现在大家越来越依靠LCMS
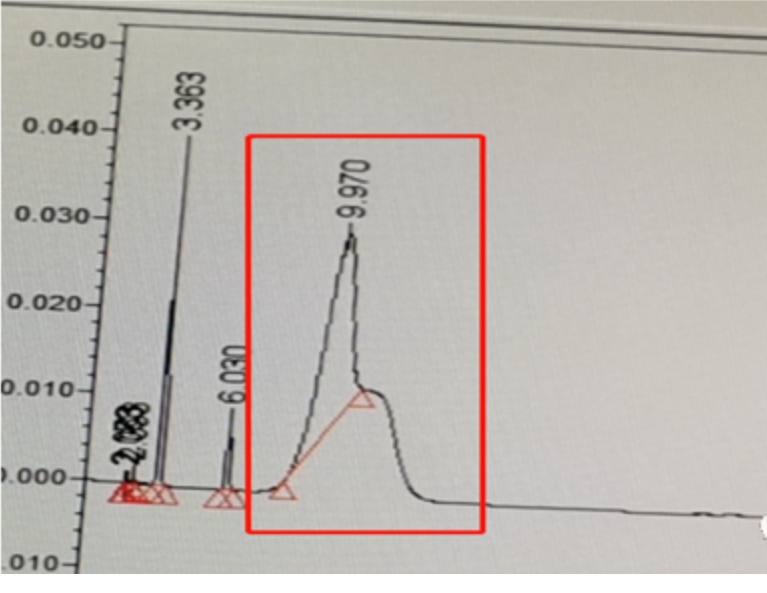
下图红框的已知杂质峰形较差从峰形上判断:1、可能是溶剂效应;2、也有可能是结构互变;以及按碱或高温改善羰基类的马鞍峰或前沿峰一文,升高柱温没有改善。如上述操作没有改善,再看一下杂质结构为一对映异构体,见下图,峰形差可能是这一对映异构体在反相中没有完全重叠,也即C18可以识别这组对映异构体。使用数据库hplcc

1.前言现在大家一致认为分子的手性是生命过程中必不可少的部分,还一致同意大多数控制活生物体生理机能的生物活性化合物都是具有手性的。因而,在针对生物活性化合物以及天然产物的结构研究上,其绝对构型的测定成为首要的重大问题。其次是成为医药目标的生物活性化合物和天然产物的手性合成,以及如何有效的利用100%对映体

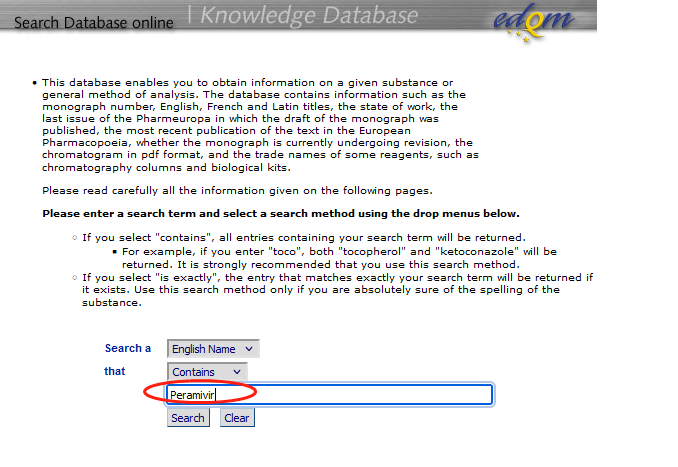
分析方法如果能直接引用药典则是最省事的。已说明每种C18色谱柱都不完全相同。药典方法无法重现的多数原因是没有正确选择色谱柱,EP和USP的个论中虽然没有提供色谱柱的品牌信息,但是在官网上提供了色谱柱的品牌信息。1、EP色谱柱检索链接https://www.edqm.eu/en/knowledge-database点击SearchKnowledge Databa

我们知道了FDA、EMA和PDMA,澳大利亚也有原研审评报告。那么澳大利亚的原研审评报告无法下载,以下是正确的打开方式:方法一澳大利亚官网https://www.tga.gov.au/选择下图红框菜单选择Industry » Prescription medicines »Regulatory decisions and notices进入以下界面,选择Australian Public Assess

药物合成工艺路线中,所有潜在杂质和实际存在的杂质,包括原料、起始物料、中间体、试剂、催化剂、溶剂、副产物、降解产物,以及可能存在的潜在杂质,例如磺酸和醇类反应的杂质,位置异构体等。切记:路线设计和试剂筛选时,要考虑到可能产生的致突变性或致癌性杂质。评估方式毒性杂质评估方式一般包括:文献检索、QSAR评估
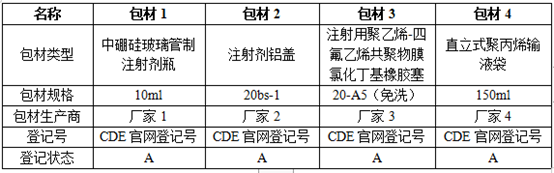
注射剂包装系统无质量风险是注射剂产品质量以及安全性合格的重要前提,那么这部分研究思路是怎样的,如何撰写申报资料以达到CDE审评要求呢?本文将对包装系统研究思路以及资料撰写思路进行讨论。包装系统研究主要有包装系统基本信息获取、包装系统的选择及匹配性、包材性能的对比以及包材相容性四个部分组成,由于篇幅有限本



 在线客服
在线客服
 快速发布
快速发布
 文章查询
文章查询
 分析化学
分析化学
 我的消息
我的消息
 文章发表
文章发表

 回到顶部
回到顶部